









Cuprins
Scleroza tuberoasă Bourneville
Ce este ?
Scleroza tuberoasă Bourneville este o boală genetică complexă caracterizată prin dezvoltarea unei tumori benigne (necanceroase) în diferite părți ale corpului. Aceste tumori pot fi apoi localizate în piele, creier, rinichi și alte organe și țesuturi. Această patologie poate provoca, de asemenea, probleme serioase în dezvoltarea individului. Cu toate acestea, manifestările clinice și severitatea bolii variază de la pacient la pacient.
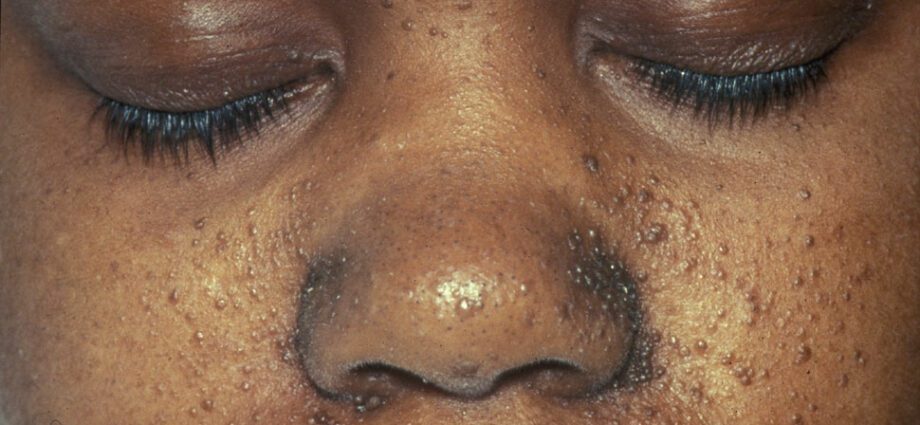
Anomaliile pielii asociate sunt în general similare cu petele de pe piele sau cu zonele în care pielea este mai deschisă decât pe restul corpului. Dezvoltarea tumorilor la nivelul feței se numește angiofibrom.
În contextul leziunilor cerebrale, semnele clinice sunt crizele de epilepsie, problemele de comportament (hiperactivitate, agresivitate, dizabilități intelectuale, probleme de învățare etc.). Unii copii cu boala au chiar o formă de autism, tulburări de dezvoltare, care afectează interacțiunile sociale și comunicarea. Tumorile benigne ale creierului pot provoca, de asemenea, complicații care pot fi fatale pentru subiect.
Dezvoltarea tumorilor la rinichi este frecventă la persoanele cu scleroză tuberoasă. Acest lucru poate provoca complicații severe ale funcției renale. În plus, tumorile se pot dezvolta în inimă, plămâni și retină. (2)
Este o boală rară, a cărei prevalență (număr de cazuri într-o anumită populație la un moment dat) se ridică la 1 / 8 până la 000 / 1 persoane. (15)
Simptome
Manifestările clinice asociate cu scleroza tuberoasă din Bourneville variază în funcție de organele afectate. În plus, simptomele asociate bolii variază foarte mult de la un individ la altul. Cu simptome de la ușoare la severe.
Cele mai identificate simptome ale acestei boli includ convulsii epileptice, tulburări cognitive și comportamentale, anomalii ale pielii etc. Organele cel mai des afectate sunt: creierul, inima, rinichii, plămânii și pielea.
Dezvoltarea de tumori maligne (canceroase) este posibilă în această boală, dar sunt rare și afectează în principal rinichii.
Semnele clinice ale bolii la nivelul creierului provin din atacuri la diferite niveluri:
– afectarea tuberculilor corticali;
– noduli ependimari (SEN);
– astrocitoame ependimale gigantice.
Acestea au ca rezultat: dezvoltarea retardului mintal, dificultăților de învățare, tulburări de comportament, agresivitate, tulburări de atenție, hiperactivitate, tulburări obsesiv-compulsive etc.
Afectarea rinichilor se caracterizează prin dezvoltarea chisturilor sau angiomiolipomilor. Acestea pot duce la dureri de rinichi și chiar la insuficiență renală. Dacă sângerarea abundentă este vizibilă, poate fi de la anemie severă sau hipertensiune arterială. Alte consecințe mai grave, dar rare pot fi, de asemenea, vizibile, în special dezvoltarea carcinoamelor (tumoare a celulelor constitutive ale epiteliului).
Leziunile oculare pot fi similare cu petele vizibile de pe retină, provocând tulburări de vedere sau chiar orbire.
Anomaliile cutanate sunt numeroase:
– macule hipomelanice: care au ca rezultat aparitia unor pete usoare pe piele, oriunde pe corp, ca urmare a unui deficit de melanina, o proteina care da culoare pielii;
– apariția petelor roșii pe față;
– pete decolorate pe frunte;
– alte anomalii ale pielii, dependente de la un individ la altul.
Leziunile pulmonare sunt prezente la 1/3 dintre pacientii cu o usoara predominanta feminina. Simptomele asociate sunt atunci dificultăți de respirație mai mult sau mai puțin severe.
Originile bolii
Originea bolii este genetică și ereditară.
Transmiterea implică mutații ale genelor TSC1 și TSC2. Aceste gene de interes intră în joc în formarea proteinelor: hamartina și tuberina. Aceste două proteine fac posibilă, printr-un joc interactiv, reglarea proliferării celulare.
Pacienții cu boală se nasc cu cel puțin o copie mutantă a acestor gene în fiecare dintre celulele lor. Aceste mutații limitează apoi formarea hamartinei sau tubertinei.
În contextul în care cele două copii ale genei sunt mutate, ele împiedică complet producerea acestor două proteine. Acest deficit proteic nu mai permite deci organismului sa regleze cresterea anumitor celule si, in acest sens, duce la dezvoltarea celulelor tumorale in diferite tesuturi si/sau organe.
Factorii de risc
Factorii de risc pentru dezvoltarea unei astfel de patologii sunt genetici.
Într-adevăr, transmiterea bolii este eficientă printr-un mod autosomal dominant. Fie, gena mutantă de interes este localizată la un cromozom non-sexual. În plus, prezența doar a uneia dintre cele două copii ale genei mutante este suficientă pentru dezvoltarea bolii.
În acest sens, o persoană care posedă unul dintre acești doi părinți care suferă de boală are un risc de 50% de a dezvolta el însuși fenotipul bolnav.
Prevenirea și tratamentul
Diagnosticul bolii este în primul rând diferențial. Se bazează pe criterii fizice atipice. În cele mai multe cazuri, primele semne caracteristice ale bolii sunt: prezența crizelor epileptice recurente și întârzierile în dezvoltarea subiectului. În alte cazuri, aceste prime semne au ca rezultat pete pe piele sau identificarea unei tumori cardiace.
În urma acestui prim diagnostic, examinările suplimentare sunt esențiale pentru a valida sau nu diagnosticul. Acestea includ:
– o scanare a creierului;
– un RMN (imagini prin rezonanță magnetică) a creierului;
– o ecografie a inimii, ficatului și rinichilor.
Diagnosticul poate fi eficient la nașterea copilului. În caz contrar, este important ca acesta să fie efectuat cât mai repede posibil pentru a prelua cât mai curând controlul pacientului.
În prezent, nu există un tratament pentru boală. Tratamentele asociate sunt deci independente de simptomele prezentate de fiecare individ.
De obicei, se administrează medicamente antiepileptice pentru a limita crizele. În plus, sunt prescrise și medicamente pentru tratamentul celulelor tumorale ale creierului și rinichilor. În contextul problemelor de comportament, este necesar un tratament specific al copilului.
Tratamentul bolii este de obicei pe termen lung. (1)