









Boala Wilson
Ce este ?
Boala Wilson este o boală genetică moștenită care împiedică eliminarea cuprului din organism. Acumularea de cupru în ficat și creier cauzează probleme hepatice sau neurologice. Prevalența bolii Wilson este foarte scăzută, în jur de 1 din 30 de persoane. (000) Există un tratament eficient pentru această boală, dar diagnosticarea ei precoce este problematică deoarece rămâne tăcută mult timp.
Simptome
Acumularea de cupru începe la naștere, dar primele simptome ale bolii Wilson nu apar adesea până la adolescență sau la vârsta adultă. Ele pot fi foarte diverse deoarece mai multe organe sunt afectate de acumularea de cupru: inima, rinichii, ochii, sângele... Primele semne sunt hepatice sau neurologice în trei sferturi din cazuri (40% și respectiv 35%), dar pot fi de asemenea să fie psihiatrice, renale, hematologice și endocrinologice. Ficatul și creierul sunt afectate în special, deoarece conțin deja cel mai mult cupru în mod natural. (2)
- Tulburări hepatice: icter, ciroză, insuficiență hepatică...
- Tulburări neurologice: depresie, tulburări de comportament, dificultăți de învățare, dificultăți de exprimare, tremor, crampe și contracturi (distonie)...
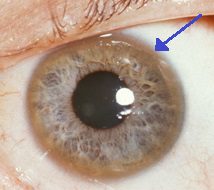
Inelul Keyser-Fleisher care înconjoară irisul este caracteristic acumulării de cupru în ochi. Pe lângă aceste simptome acute, boala Wilson se poate prezenta cu simptome necaracteristice, cum ar fi oboseală generală, dureri abdominale, vărsături și scădere în greutate, anemie și dureri articulare.
Originile bolii
La originea bolii Wilson, există o mutație a genei ATP7B situată pe cromozomul 13, care este implicată în metabolismul cuprului. Controlează producția unei proteine ATPaza 2 care joacă un rol în transportul cuprului din ficat în alte părți ale corpului. Cuprul este un element de construcție necesar pentru multe funcții celulare, dar în exces de cupru devine toxic și dăunează țesuturilor și organelor.
Factorii de risc
Transmiterea bolii Wilson este autosomal recesiv. Prin urmare, este necesar să primiți două copii ale genei mutante (de la tată și de la mamă) pentru a dezvolta boala. Aceasta înseamnă că bărbații și femeile sunt expuși în mod egal și că doi părinți purtători ai genei mutante, dar care nu sunt bolnavi, au un risc de transmitere a bolii din patru la fiecare naștere.
Prevenirea și tratamentul
Există un tratament eficient pentru a opri progresia bolii și pentru a reduce sau chiar a elimina simptomele acesteia. De asemenea, este necesar ca acesta să fie inițiat precoce, dar de multe ori sunt necesare multe luni de la apariția simptomelor pentru a diagnostica această boală tăcută, puțin cunoscută și ale cărei simptome indică multe alte afecțiuni (hepatită pentru care este afectarea ficatului și depresie pentru implicare psihiatrică) .
Un tratament „chelat” face posibilă atragerea cuprului și eliminarea acestuia prin urină, limitând astfel acumularea lui în organe. Se bazează pe D-penicilamină sau Trientină, medicamente administrate pe cale orală. Sunt eficiente, dar pot provoca reacții adverse grave (leziuni renale, reacții alergice etc.). Cand aceste reactii adverse sunt prea importante, apelam la administrarea de zinc care va limita absorbtia cuprului de catre intestine.
Un transplant de ficat poate fi necesar atunci când ficatul este prea deteriorat, ceea ce este cazul pentru 5% dintre persoanele cu boala Wilson (1).
Un test de screening genetic este oferit fraților unei persoane afectate. Dă naștere unui tratament preventiv eficient în cazul în care este detectată o anomalie genetică în gena ATP7B.