









Cuprins
Sindromul Treacher-Collins
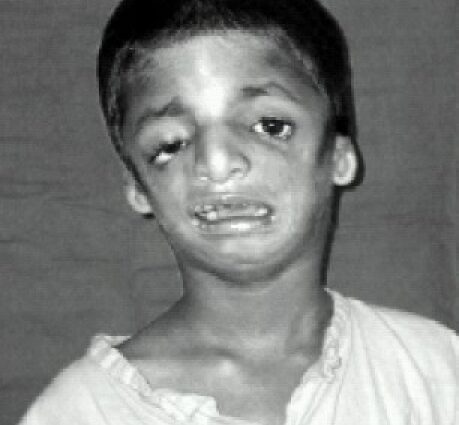
O boală genetică rară, sindromul Teacher-Collins se caracterizează prin dezvoltarea unor malformații congenitale ale craniului și feței în timpul vieții embrionare, având ca rezultat deformări ale feței, urechilor și ochilor. Consecințele estetice și funcționale sunt mai mult sau mai puțin severe și unele cazuri necesită numeroase intervenții chirurgicale. Cu toate acestea, în majoritatea cazurilor, preluarea controlului permite păstrarea unei anumite calități a vieții.
Ce este Sindromul Treacher-Collins?
Definiție
Sindromul Treacher-Collins (numit după Edward Treacher Collins, care l-a descris pentru prima dată în 1900) este o boală congenitală rară care se manifestă încă de la naștere cu malformații mai mult sau mai puțin severe ale părții inferioare a corpului. fata, ochii si urechile. Atacurile sunt bilaterale și simetrice.
Acest sindrom se mai numește și sindromul Franceschetti-Klein sau disostoză mandibulo-facială fără anomalii finale.
Cauze
Până acum se știe că trei gene sunt implicate în acest sindrom:
- gena TCOF1, situată pe cromozomul 5,
- genele POLR1C și POLR1D, localizate pe cromozomii 6 și respectiv 13.
Aceste gene direcționează producția de proteine care joacă un rol cheie în dezvoltarea embrionară a structurilor faciale. Alterarea lor prin mutații perturbă dezvoltarea structurilor osoase (în principal cele ale maxilarului inferior și superior și ale pomeților) și a țesuturilor moi (mușchii și pielea) din partea inferioară a feței în a doua lună de gestație. De asemenea, sunt afectate pinna, canalul urechii precum și structurile urechii medii (ossicule și/sau timpane).
Diagnostic
Malformațiile faciale pot fi suspectate din ecografie din trimestrul II de sarcină, mai ales în cazurile de malformații semnificative ale urechii. În acest caz, diagnosticul prenatal va fi stabilit de o echipă multidisciplinară din imagistica prin rezonanță magnetică (RMN) a fătului, permițând vizualizarea cu mai multă precizie a malformațiilor.
De cele mai multe ori, diagnosticul se pune printr-un examen fizic facut la nastere sau imediat dupa. Datorita variabilitatii mari a malformatiilor, aceasta trebuie confirmata intr-un centru specializat. Un test genetic pe o probă de sânge poate fi comandat pentru a căuta anomaliile genetice implicate.
Unele forme ușoare trec neobservate sau vor fi depistate întâmplător târziu, de exemplu în urma apariției unui nou caz în familie.
Odată pus diagnosticul, copilul este supus unei serii de examinări suplimentare:
- imagistica facială (radiografie, tomografie computerizată și RMN),
- examene de ureche și teste de auz,
- evaluarea vederii,
- căutarea apneei în somn (polisomnografie)...
Oamenii în cauză
Se crede că sindromul Treacher-Collins afectează unul din 50 de nou-născuți, atât fete, cât și băieți. Se estimează că aproximativ 000 de cazuri noi apar în fiecare an în Franța.
Factorii de risc
Consilierea genetică într-un centru de referință este recomandată pentru a evalua riscurile de transmitere genetică.
Aproximativ 60% din cazuri apar izolat: copilul este primul pacient din familie. Malformațiile apar în urma unui accident genetic care a afectat una sau alta dintre celulele reproducătoare implicate în fertilizare (mutație „de novo”). Gena mutantă va fi apoi transmisă descendenților săi, dar nu există niciun risc deosebit pentru frații săi. Cu toate acestea, ar trebui verificat dacă unul dintre părinții săi nu suferă de fapt de o formă minoră a sindromului și poartă mutația fără să știe.
În alte cazuri, boala este ereditară. Cel mai adesea, riscul de transmitere este unul din doi cu fiecare sarcină, dar în funcție de mutațiile implicate, există și alte moduri de transmitere.
Simptomele sindromului Treacher-Collins
Trăsăturile feței celor afectați sunt adesea caracteristice, cu bărbia atrofiată și în retragere, pomeți inexistenți, ochii înclinați în jos spre tâmple, urechi cu pavilion mic și prost tivituit, sau chiar complet absenți...
Principalele simptome sunt legate de malformații ale sferei ORL:
Dificultăți respiratorii
Mulți copii se nasc cu căile respiratorii superioare înguste și gura îngust deschisă, cu o cavitate bucală mică, în mare parte obstrucționată de limbă. Prin urmare, dificultăți semnificative de respirație, în special la nou-născuți și sugari, care sunt exprimate prin sforăit, apnee în somn și respirație prea slabă.
Dificultăți de a mânca
La sugari, alăptarea poate fi îngreunată de dificultăți de respirație și de anomalii ale palatului și palatului moale, uneori divizate. Hranirea este mai usoara dupa introducerea alimentelor solide, insa mestecatul poate fi dificil si problemele dentare sunt frecvente.
Surditate
Disconfortul auditiv datorat malformațiilor urechii externe sau medii este prezent în 30 până la 50% din cazuri.
Tulburări vizuale
O treime dintre copii suferă de strabism. Unii pot fi, de asemenea, miop, hipermetropi sau astigmatici.
Dificultăți de învățare și comunicare
Sindromul Treacher-Collins nu cauzează deficit intelectual, dar surditatea, problemele de vedere, dificultățile de vorbire, repercusiunile psihologice ale bolii precum și tulburările induse de îngrijiri medicale adesea foarte grele pot provoca o întârziere. limbaj și dificultăți de comunicare.
Tratamente pentru sindromul Treacher-Collins
Îngrijirea copilului
Suportul respirator și/sau alimentația cu tub pot fi necesare pentru a facilita respirația și hrănirea sugarului, uneori încă de la naștere. Când asistența respiratorie trebuie menținută în timp, se efectuează o traheotomie (mică deschidere în trahee, la nivelul gâtului) pentru introducerea directă a unei canule care asigură trecerea aerului în căile respiratorii.
Tratamentul chirurgical al malformațiilor
Intervenții chirurgicale mai mult sau mai puțin complexe și numeroase, referitoare la palatul moale, maxilare, bărbie, urechi, pleoape și nas pot fi propuse pentru a facilita alimentația, respirația sau auzul, dar și pentru a reduce impactul estetic al malformațiilor.
Cu titlu indicativ, fantele palatului moale se închid înainte de vârsta de 6 luni, primele proceduri cosmetice pe pleoape și pomeți de la 2 ani, prelungirea mandibulei (distragerea mandibulei) spre 6 sau 7 ani, reatașarea de pavilionul urechii la vârsta de 8 ani, mărirea canalelor auditive și/sau intervenția chirurgicală a oselor pe la 10 până la 12 ani... Alte operații de chirurgie estetică se mai pot face la adolescență...
Aparat auditiv
Aparatele auditive sunt uneori posibile de la vârsta de 3 sau 4 luni când surditatea afectează ambele urechi. Sunt disponibile diferite tipuri de proteze în funcție de natura leziunii, cu o eficiență bună.
Urmarire medicala si paramedicala
Pentru limitarea și prevenirea dizabilității, monitorizarea periodică este multidisciplinară și apelează la diverși specialiști:
- ORL (risc ridicat de infecție)
- Oftalmolog (corectarea tulburărilor de vedere) și ortoptist (reabilitare oculară)
- Medic stomatolog si ortodont
- Logoped…
Sprijinul psihologic și educațional este adesea necesar.